Brett Milash, Center for High Performance Computing, University of Utah
One of my roles at the University of Utah involves supporting scientists developing computing work flows on the computer clusters here at the Center for High Performance Computing. Toward this goal I am evaluating a number of the countless work flow management tools out there and creating a guide to help scientists choose which one to use. The scope of work flows can vary, from simple data processing to data download, data quality assessment, analysis, manuscript preparation, and so on, so it’s clear that one size will not fit all, and that different work flows may be better served by different work flow managers. In the end I hope to create a resource to help researchers choose the right tool for the job, with links to documentation and download sites for each tool, examples of use, quick recipes showing how to get simple things done, and “gotchas” and workarounds.
My evaluation project started with the selection a test work flow. This test case needed to:
- be sufficiently complex, i.e., not a trivial task
- require cluster computing, and also local tasks
- require both serial and parallel jobs
- consist of multiple steps
- be familiar to me, so I can focus on the work flow manager rather than test case itself.
I selected a bioinformatics work flow, RNAseq gene expression analysis, since it is very familiar to me and it meets all the other criteria. An added bonus is that it is a work flow required by some of the scientists I support, so I figured I could kill two birds with one work flow. The only shortcoming of this analysis is that it does not require multi-node MPI computing tasks, only single node tasks – perhaps I can engineer something to perform part of the analysis in a multi-node way.
I intend to implement my test work flow in each of the work flow managers, evaluating each manager on a long wish list of features in the context of a common work flow. My evaluation criteria include:
- Is the work flow manager general purpose or domain-specific?
- Does it use computing resources efficiently?
- Is it multithreaded?
- Does it provide cluster support?
- Does it reduce reprocessing (faulttolerant / restartable / automatic retry)?
- Can you execute a subset of the work flow?
- Is it easy to use?
- Does it require special (programming) skills?
- Is it well documented?
- Is it actively used and supported?
- Are work flows modular / reusable?
- Does it display work flow structure / dependency graph?
- Is the work flow manager open source / free to use?
- Does it support collaboration?
- Does it provide control over logging?
- Other criteria?
Candidate work flow managers include snakemake, Makeflow, Pegasus, Galaxy, Drake, Cromwell, Swift, and Apache Taverna.
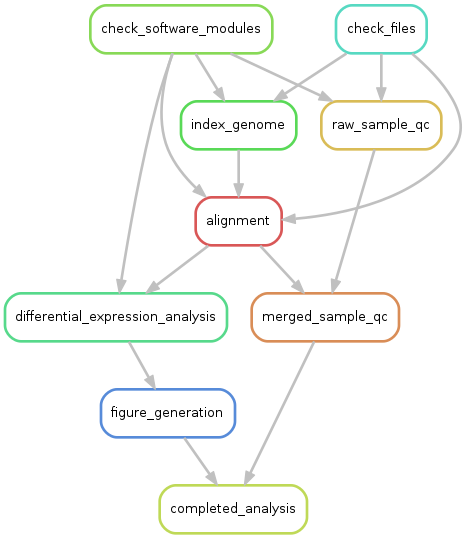
My first candidate for evaluation was snakemake ( http://snakemake.readthedocs.io/en/stable/index.html ). This is a work flow tool from Johannes Koster and Sven Rahmann at the University of Duisburg-Essen. Although it comes from the bioinformatics world, snakemake is in no way bioinformatics-specific; it is a general-purpose tool. Installation was simple, and the example-filled documentation is clear and concise. Snakemake represents a work flow as a collection of inter-connected rules, each rule controlling one step of the work flow. When I started construction of my test work flow, I was at first puzzled whether I should think of rules as verbs or nouns, whether the rules were the actions or actors in the work flow. Now, I think of the rules as complete sentences, like “check the quality of one sample’s data”, or “align a fastq file against the genome index”. These sentences contain the input, the action to be taken, and at least imply what the output will be. After a few very minor setbacks I completed the work flow using this excellent work flow manager, and wrote up the results here: https://wiki.chpc.utah.edu/display/~u0424091/Snakemake .
I also made a few general work flow discoveries during my evaluation of snakemake. It is helpful to start a work flow by checking for all the prerequisites, whether they are data files or installed software. I learned the value of having a very small test data set, one that can be processed locally, and one whose processing would succeed or fail very quickly. I also learned a useful technique for work flow development: one can create the skeleton of a work flow to define the shape of the problem before doing any data processing. Once the skeleton is in place, you can flesh out the rules with actual data processing commands, testing as you go.
I’m looking forward to repeating this process with other work flow managers, to identify their strengths and weaknesses and to see how they compare with snakemake.
References:
Snakemake—a scalable bioinformatics workflow engine
Johannes Köster Sven Rahmann
Bioinformatics, Volume 28, Issue 19, 1 October 2012, Pages 2520–2522, https://doi.org/10.1093/bioinformatics/bts480